|
Konsekwencje poznania genomów mikroorganizmów
Pełna sekwencja pierwszych dwóch genomów bakteryjnych Mycoplasma genitalium i Haemophilus influenzae, została ogłoszona w 1995 r. przez prywatną firmę C. Ventera, TIGR (The Institute of Genomic Research). W ciągu następnych lat zsekwencjonowano dalszych 50 genomów i około 130 jest w trakcie sekwencjonowania. Dyscyplina biologii skupiająca się na systematycznym badaniu genomów to genomika strukuralna i funkcjonalna. Z genomiki wywodzą się bezpośrednio proteomika, czyli nauka o wszystkich białkach syntetyzowanych w organizmie oraz transkryptomika, której celem jest badanie ilościowe i jakościowe informacyjnego RNA.
Większość zsekwencjonowanych bakterii to patogeny ludzi i zwierząt. Uzasadnieniem dla podjęcia wysiłku eksperymentalnego i finansowego była więc nadzieja poznania mechanizmów patogenezy oraz możliwość opracowania nowych leków i szczepionek. Część zsekwencjonowanych genomów należy do archebakterii i bakterii o szczególnie ciekawym metabolizmie. Deinococcus radiodurans zadziwia szczególną odpornością na promieniowanie gamma (Science 1999, 286, 1571). Dawki milionów radów nie wpływają ani na przeżywalność tych bakterii ani na poziom mutacji. Spodziewano się więc odkrycia szczególnie wydajnych systemów naprawczych i rzeczywiście takie systemy znaleziono, często w wielu powtórzeniach. Aeropyrum pernix (DNA Res. 1999, 6, 83), Aquifex aeolicus (Nature 1998, 392, 353) i Archeoglobus fulgidus (Nature 1997, 390, 364) należą do tzw ekstremofili, o optymalnej temperaturze wzrostu 85-95oC, interesująca więc była odpowiedź na pytanie jakie białka i inne struktury ochronne tworzą takie bakterie. Sekwencja genomu obligatoryjnego symbionta mszyc Buchnera sp. wykazała, że to genom bakterii koduje aminokwasy niezbędne dla metabolizmu mszyc (Nature 2000, 407, 81). Osiągnięciem ostatnich miesięcy jest podanie pełnej sekwencji Pseudomonas aeruginosa oportunistycznego patogena w mukowiscydozie, ale też bakterii powszechnie spotykanej w różnych środowiskach (Nature 2000, 406, 959). Jest to największy do tej pory zsekwencjonowany genom bakterii (5 570 otwartych ramek odczytu, 6.3 Mpz), z ogromną ilością genów regulatorowych, rozbudowanym katabolizmem i licznymi genami oporności na antybiotyki. Poznana do tej pory sekwencja różnych genomów wyjaśniła przede wszystkim ich organizację. Mitem okazała się hipoteza kolistości chromosomów bakteryjnych. Genom bakteryjny mogą stanowić zarówno koliste jak i liniowe chromosomy i plazmidy. Rekord bije Borrelia burgdorferi czynnik sprawczy boreliozy, która obok liniowego chromosomu ma 21 liniowych i kolistych plazmidów (Nature 1997, 390, 580). Jak można się było spodziewać poznanie sekwencji DNA nie odpowiedziało na wszystkie postawione pytania i potrzebne są dalsze badania zmierzające do poznania funkcji genów.
Poznanie sekwencji genomów bakteryjnych oraz ogromny postęp bioinformatyki porządkującej dane pozwolił na porównanie całych genomów. Analiza komputerowa genomów prokariotycznych wykazała, że 70% białek ma domeny konserwatywne. Biorąc za podstawę różne domeny konserwatywne białek z 21 zsekwencjonowanych genomów zgrupowano białka w grupy pokrewne funkcjonalnie i filogenetycznie (Nucl. Acids Res. 2000, 28, 33). Utworzono ponad 2000 grup tzw. COGs (cluster of ortologous groups), które stanowią 56-86% produktów białkowych zsekwencjonowanych genomów. Ta baza danych jest skojarzona z programem pozwalającym na dopasowanie nowego białka do określonej grupy ortologów. Można w ten sposób przewidzieć położenie filogenetyczne i funkcję nowego, nieznanego białka. Baza danych zawiera też grupy białek paralogicznych powstałych przez duplikację w obrębie jednego genomu, spełniających podobne funkcje. Im prostszy organizm tym więcej jego genów jest ortologami tzn. używa w metabolizmie bardziej konserwatywnych białek. Ortologi są też podzielone na 17 grup funkcjonalnych i co ciekawe największą grupę stanowią białka o zupełnie nieznanej funkcji.
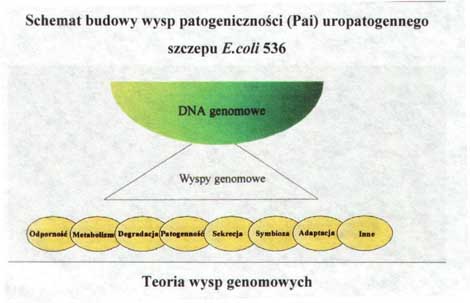
Poznanie genomów kilkudziesięciu mikroorganizmów dostarczyło dowodu na ich molekularną ewolucję. Można było podać molekularny dowód na istnienie horyzontalnego transferu genów, czyli międzygatunkowego, jednoetapowego przeniesienia grup genów pozwalającego na adaptację bakterii do nowego środowiska (Genome Res. 10, 1719). Około 1.5%-14.5% genów Archea i Bacteria została nabyta w wyniku horyzontalnego transferu genów poprzez transformację, koniugację i transdukcję. Zidentyfikowano tzw. wyspy patogeniczności, czyli duże (10-200 kpz) regiony genomu skupiające geny dla czynników wirulencji (adhezyny, toksyny, inwazyny, systemy sekrecji białek, systemy pobierania żelaza i in.) (Cell 1996, 87, 791). Wyspy symbiotyczne są to duże (500 kpz) regiony genomu zawierające grupy genów odpowiedzialne za symbiozę Mesorhizobium z roślinami motylkowatymi. Wyspy adaptacyjne, czyli regiony genomu skupiające geny pozwalające na przeżywanie w określonym środowisku (np. Pseudomonas putida rozkładający chlorokatechole). Wyspy genomowe są rozpoznawane w genomie na podstawie odmiennej zawartości par GC w stosunku do całego genomu, odmiennej używalności kodonów oraz obecności genów i sekwencji świadczących o ruchliwości wysp, np. genów dla transposaz, integraz fagowych, rekombinaz, końcowych powtórzeń, czy elementów IS. Ewolucja mikroorganizmów według teorii wysp genomowych przebiegałaby nie poprzez stopniowe gromadzenie mutacji punktowych w genomie, a poprzez nabywanie fragmentów DNA umożliwiających przystosowanie mikroorganizmów do różnych nisz ekologicznych (Rys. 1).
Komputerowa analiza sekwencji zasad genomu Escherichia coli umożliwiła również badania, które nazwano molekularną archeologią (Proc. Natl. Acad. Sci., 1998, 95, 9413). Można zbadać ewolucyjną historię genów na podstawie oszacowania stopnia transferu poziomego genów oraz tzw. współczynnika upodobnienia (ang. amelioration) co jest funkcją względnego tempa mutacji G\C do A\T. Stwierdzono, że około 100 mln lat temu dwa gatunki E. coli i Salmonella enterica oddzieliły się od siebie w wyniku 234 przypadków horyzontalnego transferu aż 18% genów (755 spośród 4288). Średni wiek wprowadzonych genów określa się na 14.4 mln lat.
Sekwencjonowanie genomów mikroorganizmów i organizmów wyższych ma ogromne znaczenie dla nauk podstawowych, ale też dla medycyny, diagnostyki medycznej, farmakologii, terapii genowej i wielu innych dziedzin. Można też obserwować gwałtowny rozwój technologii instrumentalnej i bioinformatyki zajmującej się zbieraniem, analizą i interpretacją ogromnej ilości danych tworzonych w wyniku postępu genomiki.
Anna Skorupska
Prof. dr hab. Anna Skorupska pracuje w Zakładzie Mikrobiologii Ogólnej, Wydziału BiNoZ UMCS w Lublinie

powrót do wydawnictwa 
|
